





New Delhi: Kerala was held up as the model state in the early months of the Covid-19 pandemic, as it was able to efficiently trace and isolate people who were infected with the SARS-CoV-2 virus.
But as the lockdown was eased and inter-state travel resumed, Kerala began struggling to manage the spread of Covid-19.
Genetic sequencing of over 4,000 samples of the novel coronavirus has now revealed how the virus crossed into Kerala from multiple states, causing a second wave that is yet to peak. There are many more insights it has provided — like confirming incidents of reinfection and identifying a cluster of virus unique to India.
On 31 March, just six days into the lockdown, ThePrint had busted claims that the Covid-19 strain in India is a less virulent mutation than the one in other countries. At the time, only two samples had been sequenced, too few for scientists to draw any conclusions about the nature of the virus in India.
Even now, there is no conclusive proof to show that the virus mutations are making it more infectious.
Researchers from the Centre for Scientific and Industrial Research’s (CSIR) Institute of Genomics and Integrative Biology (IGIB) have now collated data from several labs across India to paint an accurate picture of how the pandemic spread in the country.
Also read: Scientists dissect the genetic architecture of SARS-CoV-2, to understand how it replicates
What is genetic sequencing and why is it required?
Every living organism is run by a predefined set of genes that ‘instruct’ cells how to build proteins. Understanding the sequence of genes is thus like cracking the code to the organism and how it functions.
The genetic material of the coronavirus is ribonucleic acid (RNA) strands. Each virus has about 26,000 to 32,000 bases or RNA “letters” in its length.
These letters — A, C, U and G — stand for adenine, cytosine, uracil and guanine, nitrogen-containing biological molecules that are the fundamental units of the genetic code. How A, C, U and G are arranged in the genetic code determines what proteins are expressed by the organism.
When a virus multiplies inside the cells of a living organism, it creates copies of the RNA. However, the process it uses to make these copies is not perfect, and often introduces tiny errors in the sequence of bases. These errors are called genetic mutations.
Mutations that do not help the survival of the virus eventually get eliminated, while others keep getting copied in the next generation of the virus.
By tracking these changes, scientists can visualise how the virus is evolving. Unravelling the genetic code, letter-by-letter, is known as genetic sequencing. The technique not only reveals how the virus functions, but by following the proverbial ‘breadcrumbs’ of errors, scientists can trace the path the virus took to enter a region.
Genetic sequencing also reveals how the virus originated, and to some extent, help predict how it may change in the future. For example, using computer simulations, a team from Shillong’s North-Eastern Hill University, Japan’s RIKEN Center for Biosystems Dynamics Research, and the Solan-based Jaypee University of Information Technology predicted that SARS-CoV-2 may develop resistance to the drug remdesivir — the first drug to be approved for Covid-19 treatment in the US.
Genetic sequencing is also a valuable tool for vaccine development — the Moderna Covid vaccine candidate is made of ‘messenger RNA (mRNA)’, which mimics specific parts of the genetic codes of the virus. The genetic material triggers the production of protein that tricks the body into thinking that it has encountered a foreign pathogen, and hence, launching an immune response.
Types of mutations
Although the genetic code is fundamental to how an organism functions, not all mutations in the SARS-CoV-2 translate into changes in the virus.
The genetic code is read in sets of three letters, together known as codons. Each set of three letters ‘translates’ into an amino acid, which are compounds that form the building blocks of proteins. The following chart explains how the bases are translated into amino acids.
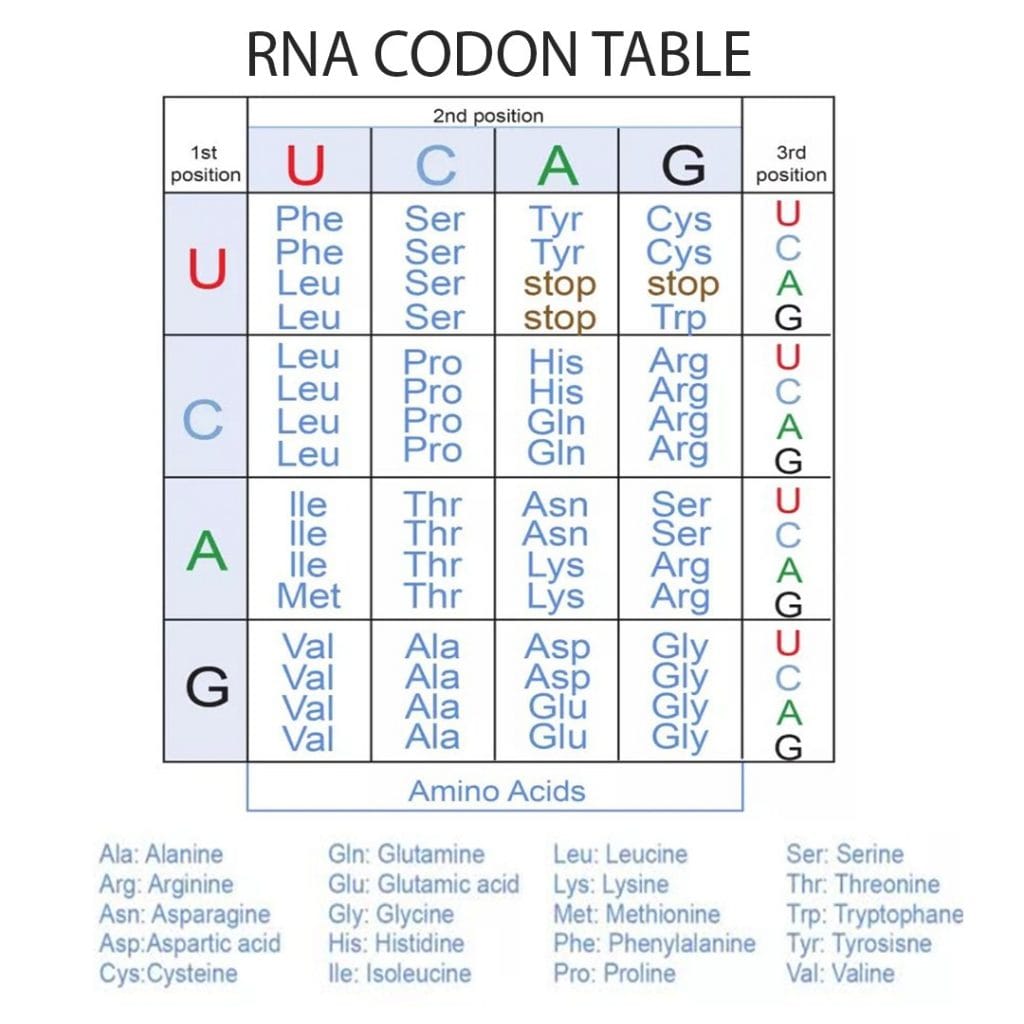
There are several types of mutations, broadly categorised as point mutations and frame-shift mutations.
Point mutations refer to when a single letter gets substituted in the genome sequence. This can be further divided into three types.
The first, known as a ‘silent’ mutation, is when a change in the base of the genetic code makes no difference when translated. For example, if the sequence changes from CUU to CUC, the amino acid expressed will remain leucine.
The second type is known as a ‘missense’ mutation. In this case, while the amino acid changes, it may or may not have a significant impact on the nature of the virus. It all depends on what effect the amino acid has on the protein.
The third type of point mutation is known as a ‘nonsense’ mutation, which basically signals the biological processes to stop translation. This results in a short, possibly dysfunctional protein. An example of this would be if the sequence UAC changed to UAA.
Frameshift mutations have more drastic effects. These occur when a base gets added into or deleted from the genome sequence. Since the genetic code is read in sets of three, everything after the point of addition or deletion ends up translating into a different amino acid, thereby completely changing the resulting proteins.
For example, consider this gene sequence: GCU ACG GAG CUU CGG. This sequence will translate into the following sequence of amino acids: Alaline, Theonine, Glutamate, Leucine, Arginine.
However, if the A at the fourth position in the above sequence gets deleted, the sequence shifts one letter to the left, and changes to GCU CGG AGC UUC GG. The first codon will still translate to Alaline, but the rest of the codons will change, translating to a completely different set of amino acids: Alaline, Arginine. Serine, Phenylalaline.
As a result, the proteins expressed by the gene sequences will be completely different with just one deletion.
Also read: Fauci says new mutation may speed the spread of coronavirus
Mutations found in India
The mutations identified by sequencing thousands of SARS-CoV-2 samples from across the world have been classified into at least 10 ‘clades’.
“The clade A2a, which is also the predominant clade in rest of the countries, is the most prevalent clade in India, comprising over 70 per cent of the genomes sequenced till date,” Bani Jolly, PhD scholar at New Delhi’s CSIR-IGIB, told ThePrint.
Jolly explained that this is defined by two mutations. One is D614G, which is a mutation in the spike protein of the virus, a protein that is known to play a role in host infection. The name D614G encodes the names of the amino acids at the 614th position. This particular mutation changes Aspartic acid (D) at the 614th position to Glycine (G).
The other mutation is P314L, in what is known as the ‘Orf1b’ gene. Here, the amino acid proline (P) at the 314th position changes to leucine (L).
“The second most prevalent clade in India is what we have named in our study as clade I/A3i. This clade comprised about 20 per cent of the genomes sequenced from India,” Jolly explained.
Four mutations define this clade — A88V in the Orf1b gene, P13L in the nucleocapsid protein, T2016K in the Orf1a and C23929T in the Orf1a.
Most genomes in this clade also share the mutation L3606F in the Orf1a, which is a characteristic mutation for another clade, A3.
Interestingly, the I/A3i clade was found to be present in distinctly large numbers in India, with limited representation outside the country. Delhi, Maharashtra and Telangana — where the Covid-19 spread occurred early — are among the 15 states and union territories from where this clade was found.
What does genetic sequencing reveal in Indian context?
The first example of the I/A3i clade in India was sampled on 16 March from an Indonesian traveller in Telangana.
A study that described this cluster in detail suggested that this clade was introduced at a single point in the country around February, following which the mutation spread countrywide, but mostly affecting the southern states. This clade thus represents a super-spreader event that led to a large outbreak of cases.
News reports from 16 March suggest that a group of Indonesian travellers participated in a religious event organised by the Tablighi Jamaat in Delhi’s Nizamuddin that turned into a super-spreader event.
Members of the group were later booked for violation of visa rules, cheating and disobedience of official orders, which aided the spread of the disease in Telangana.
“Our analysis suggests that the Clade I/A3i was represented in almost all states from which genomes are available. Members of the Clade I/A3i formed the predominant class of isolates from the states of Delhi, Telangana, Maharashtra, Karnataka and Tamil Nadu and the second largest in membership in Haryana, Madhya Pradesh, West Bengal, Odisha, Uttar Pradesh and Bihar,” the study said.
Very few samples belonging to this clade have been identified elsewhere in the world. This study displays how genetic sequencing can trace the origin and spread of infectious diseases.
While in the initial months of the pandemic, most SARS-CoV-2 samples belonged to the I/A3i clade, eventually the A2a clade became predominant in India.
The A2a clade had multiple points of emergence, which means it arrived in India through multiple sources, and then spread to other regions of the country, Jolly said.
Over 70 per cent of the genomes sequenced till date in India belong to the A2a clade, which originated from Europe.
“Through our own experience of sequencing the virus using a protocol known as CovidSeq, we have proposed that whole-genome sequencing is not only beneficial in detecting the virus (as an alternative to the currently followed RT-PCR test approaches) but also helps in understanding the epidemiology of the disease in terms of its origin and transmission in a region,” added Vinod Scaria of the IGIB, who is known for sequencing the first genome in India.
Scaria’s lab has been instrumental in sequencing nearly half the SARS-CoV-2 samples currently available in the public domain.
“For instance, sequencing and analysis of 200 coronavirus genomes from Kerala helped us understand that inter-state travel, particularly through neighbouring states, could have contributed to the spread of the Covid-19 in Kerala, followed by a local spread of the infection in the state,” Scaria told ThePrint.
This was elaborated in a study which is yet to be peer-reviewed. This paper also suggests that the effectiveness of the RT-PCR test may also be compromised when dealing with the A2a clade.
“This shows that these viral sequences, coupled with additional information, can be used to identify potential infection outbreaks by identifying the diversity of the virus and determining how the virus has travelled around the country,” Scaria explained.
Also read: Indiscipline, antigen tests & possible mutation behind Delhi Covid surge — expert panel chief
Confirming reinfections
One of the key questions around the pandemic was whether Covid-19 patients who have recovered once are still susceptible to the virus. Understanding this is vital to vaccine programmes.
While there were several reports of reinfections, the nature of the tests used to identify infections made it hard for scientists to confirm these events.
The RT-PCR test, which is the current gold-standard for confirming whether a patient is infected with Covid-19, has a number of limitations. For one, it is not always accurate, often producing false negatives. The other shortcoming is that the test is not able to distinguish between live virus and viral debris — so, even if a person had recovered from Covid-19, many of them tested positive again, since the genetic material of the dead virus was still present in the blood.
Genetic sequencing allowed scientists to confirm cases of reinfections. In a pre-print paper published in September, researchers shared details of two healthcare workers who were working in a hospital’s Covid-19 ward, and were infected by the SARS-CoV-2 virus twice over a span of three months. The study was later peer-reviewed and published.
Cases of reinfections have also been reported from other countries, like the US, Hong Kong and The Netherlands. In each case, researchers used genome sequencing to confirm the cases.
The findings from the CSIR led the Government of India to admit that Covid-19 reinfections are happening, although it reiterated that this was “very, very rare”. The Indian Council of Medical Research has commissioned a study to look into the cases of reinfection in the country.
Also read: Covid reinfections are real. But here’s why you shouldn’t worry about that just yet
Are any of these mutations making the virus more infectious?
Several state authorities in India have claimed that the virus circulating in their state is more infectious. Kerala Chief Secretary Dr Vishwas Mehta had told ThePrint that the “second wave” of Covid-19 in the state may be because they are dealing with a different strain of the virus that is highly infectious.
Earlier, a scientist from Gujarat had also claimed that the state was dealing with a more virulent strain.
Jolly said there was no evidence to back such claims, but at the same time, “there also isn’t sufficient scientific evidence to determine if these mutations are functionally silent or not”.
“Some experimental studies show D614G mutation to be associated with increased infectivity and decreased shedding of the virus. While this does seem like a concern, these results cannot be directly extrapolated to the real world scenario since many factors are there in human-to-human transmission,” she said.
The D614G mutation has been of particular concern because it is located in the spike protein — the part that facilitates the entry of the virus into cells. The problem is that most vaccines are designed to recognise this spike protein. If the spike protein mutates, then most vaccines will become defunct.
“However, so far, studies have shown that the D614G mutation is not located in a domain of the spike protein that is considered important for antibody-driven immunity,” Jolly said. “This evidence makes it seem unlikely that the mutation will impact the efficacy of the vaccines currently in development.”
However, there is still limited knowledge regarding the D614G and other mutations, so they must not be left out from consideration for any vaccine or therapeutic efforts.
Apart from these, there are certain deletions — a mutation in which a part of the genome sequence gets removed — that lead to attenuation or weakening of the virus.
“The two noteworthy ones are a 29 base pair deletion in the Orf8 gene, and a 382 base pair deletion in Orf7b and Orf8 genes. These deletions were found in genomes from Singapore and other countries, and were found to be associated with a decrease in the ‘fitness’ of the virus,” said Jolly.
But many additional factors such as host-virus interaction will need to be considered to effectively prove if a mutation does increase infectivity of the virus, although currently there is no evidence to indicate this is occurring.
Also read: Study identifies gene variations that increase susceptibility to Covid-19
Very detailed and useful information. Thank you